


Next: Spherocylinders with attraction Up: Mixtures of spherocylinders Previous: Introduction
Next: Spherocylinders with attraction
Up: Mixtures of spherocylinders
Previous: Introduction
The thermodynamic perturbation theory of ref. [113] requires
as input the equation of state of the hard core reference system (in
this case, the hard spherocylinder fluid) and the free-volume fraction
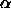 . The previous chapter (chapter 5) contains the
necessary information about the equation of state of a pure hard
spherocylinder fluid. In addition, we measured the free volume
function
. The previous chapter (chapter 5) contains the
necessary information about the equation of state of a pure hard
spherocylinder fluid. In addition, we measured the free volume
function 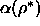 as a function of density by use of the Widom
insertion technique [36]. The advantage of the perturbation
approach is that there is no need to simulate the polymers explicitly:
it is sufficient to just measure the free volume available for a
polymer in a pure hard spherocylinder system. We should stress that
there is a slight difference in the philosophy underlying the present
(numerical) implementation of perturbation theory and the
scaled-particle theory approach of ref. [113]. Stroobants and
Lekkerkerker minimize the free energy of the polymer-colloid mixture
with respect to the orientational distribution function of the
rods. Hence, in their case, the orientational distribution function of
the mixture is not equal to the distribution function of the pure
reference system at the same density. In our simulations, we do not
attempt to vary the orientational distribution function independently
of density (as this would involve performing many simulations of a
hard spherocylinder fluid in a magnetic field).
as a function of density by use of the Widom
insertion technique [36]. The advantage of the perturbation
approach is that there is no need to simulate the polymers explicitly:
it is sufficient to just measure the free volume available for a
polymer in a pure hard spherocylinder system. We should stress that
there is a slight difference in the philosophy underlying the present
(numerical) implementation of perturbation theory and the
scaled-particle theory approach of ref. [113]. Stroobants and
Lekkerkerker minimize the free energy of the polymer-colloid mixture
with respect to the orientational distribution function of the
rods. Hence, in their case, the orientational distribution function of
the mixture is not equal to the distribution function of the pure
reference system at the same density. In our simulations, we do not
attempt to vary the orientational distribution function independently
of density (as this would involve performing many simulations of a
hard spherocylinder fluid in a magnetic field).
In the Widom method, we attempt to insert polymers (represented by
hard spheres) at random positions in
the simulation box. The fraction of insertions that does not result
in an overlap corresponds to the free volume fraction.
In practice, we measure the distance r to the nearest surface of a
spherocylinder. A polymer with a radius larger than r will
overlap with the spherocylinder, while a smaller polymer will
fit. We made a histogram of the insertion probability, and hence the
free volume fraction 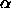 , as a function of
, as a function of 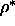 and q.
We fitted this free volume data to a polynomial in
and q.
We fitted this free volume data to a polynomial in 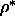 and q and used equations
1.19 and 1.20 in combination with
the simulation results for
and q and used equations
1.19 and 1.20 in combination with
the simulation results for 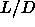 =5 (chapter 5)
to calculate the two-phase coexistence curves.
The resulting phase diagrams in the
=5 (chapter 5)
to calculate the two-phase coexistence curves.
The resulting phase diagrams in the 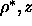 plane are presented
in figure 6.2 for q= 0.15, 0.25, 0.50, 0.65, 0.75 and
1.0. These figures show the thermodynamically stable regions of the
isotropic,
nematic, smectic A and solid phase denoted respectively by I,N,SmA and
S respectively. The phases are separated from each other by
first-order coexistence regions indicated by a grey area. Two
coexisting phases will have equal z, because z is the
fugacity of the polymer reservoir that is in equilibrium with both
phases.
In figure 6.2 a) the phase diagram for q=1.00 is depicted.
The isotropic fluid phase contains the familiar
binodal curve ending in a critical point. This binodal is very
similar to the ones we obtained in chapter 2 and
3. At a fugacity higher than
plane are presented
in figure 6.2 for q= 0.15, 0.25, 0.50, 0.65, 0.75 and
1.0. These figures show the thermodynamically stable regions of the
isotropic,
nematic, smectic A and solid phase denoted respectively by I,N,SmA and
S respectively. The phases are separated from each other by
first-order coexistence regions indicated by a grey area. Two
coexisting phases will have equal z, because z is the
fugacity of the polymer reservoir that is in equilibrium with both
phases.
In figure 6.2 a) the phase diagram for q=1.00 is depicted.
The isotropic fluid phase contains the familiar
binodal curve ending in a critical point. This binodal is very
similar to the ones we obtained in chapter 2 and
3. At a fugacity higher than 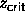 =0.95 a phase
separation occurs between a vapor phase with a low density of
spherocylinders, and
a high density liquid
phase. Upon increasing the
fugacity of the polymers, the liquid becomes metastable with
respect to the nematic
phase and we find
gas-nematic coexistence. At still higher fugacity the nematic phase
also disappears and there is a two phase gas-smectic coexistence region.
Eventually, at z=2.5 the gas-smectic is preempted by the
crystallization transition.
The important difference with the theoretical predictions of
figure 6.2 is that the scaled-particle theory of
ref. [113] does not consider the smectic and solid phases.
We find that the densities of the coexisting phases at the I-N, N-SmA
and the SmA-S transitions,
hardly change with the polymer fugacity.
=0.95 a phase
separation occurs between a vapor phase with a low density of
spherocylinders, and
a high density liquid
phase. Upon increasing the
fugacity of the polymers, the liquid becomes metastable with
respect to the nematic
phase and we find
gas-nematic coexistence. At still higher fugacity the nematic phase
also disappears and there is a two phase gas-smectic coexistence region.
Eventually, at z=2.5 the gas-smectic is preempted by the
crystallization transition.
The important difference with the theoretical predictions of
figure 6.2 is that the scaled-particle theory of
ref. [113] does not consider the smectic and solid phases.
We find that the densities of the coexisting phases at the I-N, N-SmA
and the SmA-S transitions,
hardly change with the polymer fugacity.
In figure 6.2 b) we consider the situation for a smaller polymer radius of q=0.75. As expected, the shorter interaction range results in a shift of the critical point of the I-I demixing transition to higher densities. Moreover, the transition occurs at higher polymer fugacity. We note that the I-N coexistence density gap widens (slightly) at higher polymer fugacity. The density region where the nematic phase is stable does, therefore, decrease at these high polymer fugacity. The I-I-N triple point occurs at somewhat higher fugacity. The I-N-SmA and I-SmA-S triple points are hardly affected.
When we decrease the range of interaction to q=0.65, the critical point of the I-I demixing curve disappears. The I-I binodal has moved to higher fugacity where the phase transition is preempted by the (successively) I-N, I-SmA and I-S transitions. The I-N-SmA and I-SmA-S triple points still occur at roughly the same polymer fugacity, although close to the triple points, all three first order phase coexistence regions widen somewhat.
For q=0.50 the widening of the N-SmA transition becomes very
pronounced for z>1.5. The I-N-SmA triple point increases in fugacity
faster than the N-SmA-S triple point. This shift in the relative
location of triple points continues and for q=0.25 the N-SmA-S
occurs at lower z than the I-N-S triple point. The interaction range
is now small enough to affect the SmA-S transition. The shape of the
coexistence curve in the solid phase is typical of a system
approaching an isostructural solid-solid transition. However, the
interaction ranges is still too large to induce such a phase separation
inside the solid phase. As we saw in chapter 3, in the
case of spherical colloids this transition occurs only if the
interaction range  is less than 0.07. The last plot of
figure 6.2 shows the phase diagram for q=0.15. The SmA-S
transition has become even wider, and the I-N-S and the N-SmA-S triple
points both move to higher z-values, but their relative position is
hardly affected.
is less than 0.07. The last plot of
figure 6.2 shows the phase diagram for q=0.15. The SmA-S
transition has become even wider, and the I-N-S and the N-SmA-S triple
points both move to higher z-values, but their relative position is
hardly affected.
Summarizing, the global picture that follows from this perturbation
theory calculation is as follows. For large polymers we have
an isotropic fluid-fluid phase separation ending in a critical point which
shifts to larger z and 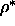 and is preempted when the radius
becomes smaller then q=0.6. The I-N-SmA and I-SmA-S triple points
occur at z=2.2 and 2.5 respectively and are not very sensitive to
the polymer size for q>0.6. For lower q both triple points move to higher
z, but the I-N-SmA overtakes the I-SmA-S triple point. For
q<0.45 the situation changes and we have and I-N-S and a N-SmA-S
triple point. For the low q values the I-N, N-SmA and SmA-S
coexistence regions fan out for high z. We find no evidence for an N-N
transition, as predicted by the
SPT approach of ref. [113].
In particular, for q=0.1 the scaled-particle theory predicts a N-N critical
point at
and is preempted when the radius
becomes smaller then q=0.6. The I-N-SmA and I-SmA-S triple points
occur at z=2.2 and 2.5 respectively and are not very sensitive to
the polymer size for q>0.6. For lower q both triple points move to higher
z, but the I-N-SmA overtakes the I-SmA-S triple point. For
q<0.45 the situation changes and we have and I-N-S and a N-SmA-S
triple point. For the low q values the I-N, N-SmA and SmA-S
coexistence regions fan out for high z. We find no evidence for an N-N
transition, as predicted by the
SPT approach of ref. [113].
In particular, for q=0.1 the scaled-particle theory predicts a N-N critical
point at 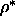 =0.6. However, the present simulations suggest
that, at that density, the nematic phase is no
longer stable with respect to the smectic.
The perturbation theory that we use becomes less accurate at
high z, because it neglects any structural changes induced by the
polymer. Still, this limitation is unlikely to affect our conclusion
that a polymer-induced N-N phase
separation will not occur for rods with an aspect ratio
=0.6. However, the present simulations suggest
that, at that density, the nematic phase is no
longer stable with respect to the smectic.
The perturbation theory that we use becomes less accurate at
high z, because it neglects any structural changes induced by the
polymer. Still, this limitation is unlikely to affect our conclusion
that a polymer-induced N-N phase
separation will not occur for rods with an aspect ratio 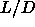 =5. After all, the density range over which the nematic
phase is stable, is quite limited: there is hardly space for an N-N
binodal. We therefore expect a
nematic-nematic phase separation to take place for larger elongation
of the spherocylinder, where the nematic phase
extends over a much wider density range.
=5. After all, the density range over which the nematic
phase is stable, is quite limited: there is hardly space for an N-N
binodal. We therefore expect a
nematic-nematic phase separation to take place for larger elongation
of the spherocylinder, where the nematic phase
extends over a much wider density range.
For the same reason, it is unlikely that there is a smectic to smectic
transition for 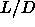 =5. Moreover, increasing the aspect ratio of the
rods will do little to facilitate the occurrence of a smectic-smectic
transition, as the density range where the smectic phase is stable
hardly changes with
=5. Moreover, increasing the aspect ratio of the
rods will do little to facilitate the occurrence of a smectic-smectic
transition, as the density range where the smectic phase is stable
hardly changes with 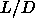 . It is tempting to speculate that
polydispersity of the rods may change these conclusions:
polydispersity in the length of the rods enhances the nematic
stability, thus making a N-N separation more likely. In contrast,
polydispersity in the diameter of the spherocylinders widens the
smectic range [118]: this may favor the occurrence of a
demixing transition in the smectic phase.
. It is tempting to speculate that
polydispersity of the rods may change these conclusions:
polydispersity in the length of the rods enhances the nematic
stability, thus making a N-N separation more likely. In contrast,
polydispersity in the diameter of the spherocylinders widens the
smectic range [118]: this may favor the occurrence of a
demixing transition in the smectic phase.
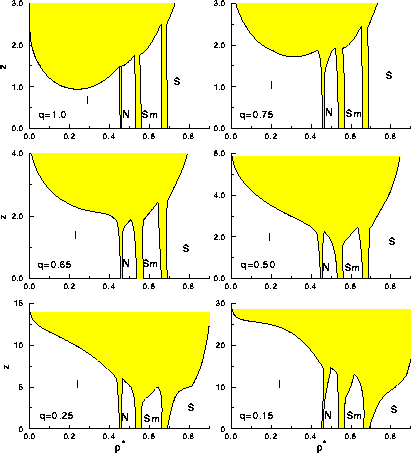
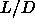 =5 mixed with polymers of different
diameter ratio q, as predicted by thermodynamic perturbation
theory. The reference system is the hard spherocylinder model, and
the perturbation was evaluated by numerical simulation of the
reference system.
=5 mixed with polymers of different
diameter ratio q, as predicted by thermodynamic perturbation
theory. The reference system is the hard spherocylinder model, and
the perturbation was evaluated by numerical simulation of the
reference system.