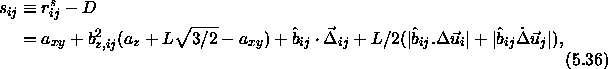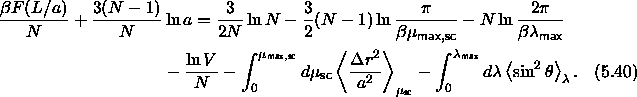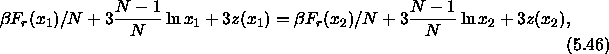Next: Phase diagram in Up: The rotator phase Previous: Finite densities
Next: Phase diagram in
Up: The rotator phase
Previous: Finite densities
The rotator solid transition is expected to be first order even in the
limit 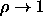 and
and 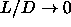 . We cannot
access this region using ordinary simulation methods.
However, somewhat surprisingly, it is possible to perform simulation
in this limit and get useful information about the limiting behavior
of the solid-solid transition.
. We cannot
access this region using ordinary simulation methods.
However, somewhat surprisingly, it is possible to perform simulation
in this limit and get useful information about the limiting behavior
of the solid-solid transition.
To see how this is achieved, let us first consider the case where
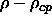 and
and 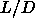 are both small but
finite. In the dense solid phase, the particles are constrained to
move in the vicinity of their lattice positions and we can safely
ignore diffusion. Therefore, we only
have to take the deviations from the lattice positions into account.
The overlap criterion of two spherocylinders i and j is based on
the shortest distance between the two cylinder axes with orientation
are both small but
finite. In the dense solid phase, the particles are constrained to
move in the vicinity of their lattice positions and we can safely
ignore diffusion. Therefore, we only
have to take the deviations from the lattice positions into account.
The overlap criterion of two spherocylinders i and j is based on
the shortest distance between the two cylinder axes with orientation
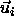 and
and 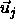 (see [77])
(see [77])

where 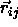 is the shortest distance vector,
is the shortest distance vector, 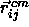 is the center-of-mass separation of neighboring particles j
and
is the center-of-mass separation of neighboring particles j
and 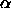 and
and 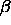 are the distances along
the axis of the two particles from the center of mass to the point
of closest approach.
In a dense solid, we can rewrite the center-of-mass separation as the vector
sum of
are the distances along
the axis of the two particles from the center of mass to the point
of closest approach.
In a dense solid, we can rewrite the center-of-mass separation as the vector
sum of 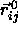 , the distance between the lattice positions of
i and j, and
, the distance between the lattice positions of
i and j, and 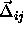 , the difference of the vector
displacements of particles i and j from their lattice positions. Hence,
, the difference of the vector
displacements of particles i and j from their lattice positions. Hence,

It is important to realize that, in the limit 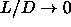 , all
spherocylinder contacts will involve only the spherical
end-caps. Moreover, near close packing, the relative particle displacements
, all
spherocylinder contacts will involve only the spherical
end-caps. Moreover, near close packing, the relative particle displacements
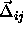 will become negligible compared to the lattice vectors
will become negligible compared to the lattice vectors
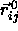 . In this limit, the vector distance of closest approach
between two spherocylinders will therefore be parallel to the lattice vector
. In this limit, the vector distance of closest approach
between two spherocylinders will therefore be parallel to the lattice vector
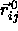 . We need therefore only consider the component of
. We need therefore only consider the component of
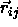 along the direction of
along the direction of 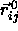
It is most convenient to express the lattice vectors 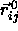 in terms of the unit vectors
in terms of the unit vectors 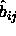 that denote the
directions of nearest neighbor bonds in the undistorted fcc
lattice. In that lattice, the lattice vector
that denote the
directions of nearest neighbor bonds in the undistorted fcc
lattice. In that lattice, the lattice vector 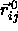 can be
written as
can be
written as

In the close-packed spherocylinder crystal, the lattice is expanded
along the 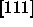 -axis by an amount
-axis by an amount 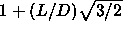 .
If we consider a crystal near (but not at) close packing, the crystal
will expand in all three directions, but not necessarily
isotropically. We assume that this expansion does not change the
symmetry of the lattice -- i.e. if we define the alignment direction
of the spherocylinders (the
.
If we consider a crystal near (but not at) close packing, the crystal
will expand in all three directions, but not necessarily
isotropically. We assume that this expansion does not change the
symmetry of the lattice -- i.e. if we define the alignment direction
of the spherocylinders (the 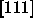 direction) to be the z-axis,
then we assume that the expanded crystal can be generated from the
close packed crystal by isotropic expansion in the xy-plane plus a
(different) expansion along the z-axis.
It is convenient to express the lattice vectors
of the expanded lattice as follows
direction) to be the z-axis,
then we assume that the expanded crystal can be generated from the
close packed crystal by isotropic expansion in the xy-plane plus a
(different) expansion along the z-axis.
It is convenient to express the lattice vectors
of the expanded lattice as follows
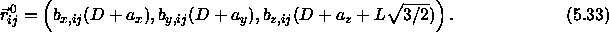
Note that, as we consider the limit 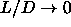 and
and
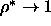 , L and
, L and 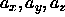 are much smaller than D.
Because of the assumed symmetry in the xy-plane,
are much smaller than D.
Because of the assumed symmetry in the xy-plane, 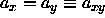 . We denote the average value of
. We denote the average value of 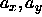 and
and
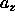 by a. It is related to the expansion of the lattice
by a. It is related to the expansion of the lattice
If we define 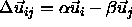 the absolute shortest distance becomes
the absolute shortest distance becomes
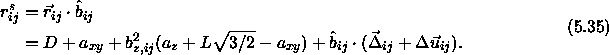
As the spherocylinders can only touch with their spherical end-caps, the surface-to-surface distance is given by
where, in the last line, we have used the values for 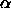 and
and
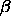 appropriate for the distance
between spherical end-caps. The spherocylinder overlap criterion in the
limit
appropriate for the distance
between spherical end-caps. The spherocylinder overlap criterion in the
limit
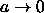 ,
, 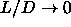 is simply
is simply

It turns out to be more convenient to express all distances in units
of a. The density enters the problem through 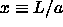 . We
can now perform a Monte Carlo simulation of this model by setting up
an undistorted fcc lattice with
unit nearest neighbor distances and move a randomly selected
sphero-cylinder i from its initial scaled displacement
. We
can now perform a Monte Carlo simulation of this model by setting up
an undistorted fcc lattice with
unit nearest neighbor distances and move a randomly selected
sphero-cylinder i from its initial scaled displacement 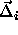 and orientation
and orientation 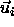 to a trial displacement and orientation in such
a way that microscopic reversibility is satisfied. We use the
conventional Metropolis rule to accept or reject the trial move.
to a trial displacement and orientation in such
a way that microscopic reversibility is satisfied. We use the
conventional Metropolis rule to accept or reject the trial move.
The system is anisotropic and we must allow box shape fluctuations to
ensure equilibrium. In the simulation the box shape is determined by
the ratio of
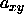 and
and 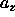 .
During trial moves that change the shape of the simulation box, the
total volume should stay constant. Equation 5.34 then implies
that the changes in
.
During trial moves that change the shape of the simulation box, the
total volume should stay constant. Equation 5.34 then implies
that the changes in 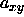 and
and 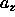 are related by
are related by

The conventional Metropolis criterion is used to decide on the acceptance of shape-changing trial moves. To speed up equilibration, we also used Molecular Dynamics simulations of the close-packed spherocylinder model. The MD scheme that we use is essentially identical to the one used in off-lattice simulations[105,106]. All the distances are scaled with a factor a. In the scaled space the new overlap criterion of eqn. 5.36 is used to locate colliding pairs. The virial expression for the pressure in the close-packing limit is
where 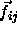 denotes the rate of momentum transfer between
sphero-cylinders i and j and the last line defines the quantity
denotes the rate of momentum transfer between
sphero-cylinders i and j and the last line defines the quantity
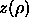 that is measured in the simulations.
Simulations were performed both by compression from ``low'' density
(i.e. low
that is measured in the simulations.
Simulations were performed both by compression from ``low'' density
(i.e. low 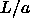 ) and by expansion from ``high'' density (high
) and by expansion from ``high'' density (high 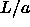 ).
The free energy of both the rotator and the aligned phases were
calculated by Einstein integration as discussed in the previous
chapter. It should be noted that the free energies of both phases do
in fact diverge at close packing. However the free energy difference is finite, and this is what we need to compute the phase
coexistence.
).
The free energy of both the rotator and the aligned phases were
calculated by Einstein integration as discussed in the previous
chapter. It should be noted that the free energies of both phases do
in fact diverge at close packing. However the free energy difference is finite, and this is what we need to compute the phase
coexistence.
In order to see how this can be achieved, consider the expression for
the free energy of a (fixed center-of-mass) Einstein crystal in the
limit of close packing. Every particle in the hard-spherocylinder
crystal is confined to move within a cell with average radius a.
We wish to switch on an Einstein spring constant that is sufficiently
large to suppress hard-core overlaps. This can only be achieved if the
spring-constant 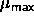 in eqn.
5.5 is of order
in eqn.
5.5 is of order
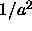 . Hence, we expect
. Hence, we expect 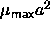 to be finite. It is
therefore convenient to write the free energy of the Einstein crystal
as follows
to be finite. It is
therefore convenient to write the free energy of the Einstein crystal
as follows
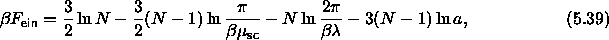
where we have defined 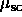 as
as 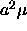 . Note that
. Note that
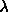 , the ``orientational'' spring constant remains finite.
The expression for the free energy of the spherocylinder crystal
(eqn.
5.4) now becomes
, the ``orientational'' spring constant remains finite.
The expression for the free energy of the spherocylinder crystal
(eqn.
5.4) now becomes
The computational scheme is essentially the same as with the
conventional Einstein crystal method.
The main difference is that all displacements in eqn.
5.40
are expressed in
units of a. The scaled coupling constant 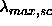 remains
finite. All divergences are now contained in the
remains
finite. All divergences are now contained in the 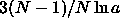 term.
When searching for the point of phase coexistence, we need to be able
to compute the variation of the free energy with density.
The free energy at any value of
term.
When searching for the point of phase coexistence, we need to be able
to compute the variation of the free energy with density.
The free energy at any value of 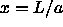 is obtained by thermodynamic
integration starting from the density
is obtained by thermodynamic
integration starting from the density 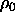 where the direct
free-energy calculation has been performed
where the direct
free-energy calculation has been performed

It is more convenient to change to the integration variable 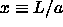 , which is related to the density through
, which is related to the density through 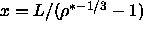 .
In the MD simulations we do not measure the pressure
.
In the MD simulations we do not measure the pressure 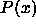 but rather
but rather 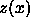 (see eqn.
5.38).
The variation of the Helmholtz free energy with x can be written as
(see eqn.
5.38).
The variation of the Helmholtz free energy with x can be written as
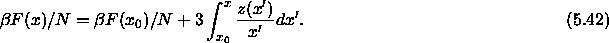
At coexistence the pressure in both phases is equal.
Using eqn.
5.38 and 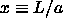 , it
is straightforward to show that the condition
, it
is straightforward to show that the condition 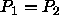 can be
written as
can be
written as
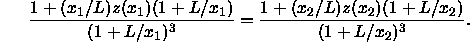
To leading order in 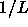 this implies that
this implies that
The chemical potential is given by

The condition for equilibrium, 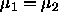 corresponds to
corresponds to
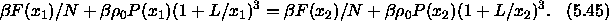
All terms in this equation diverge in the close-packing
limit. However, all differences remain finite. This is immediately
obvious for the terms involving the pressure as, at coexistence,
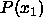 =
= 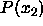 . We recall that the free energy diverges as
. We recall that the free energy diverges as
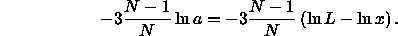
We can therefore write the Helmholtz free energy of the crystal as a
non-diverging part 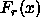 , where the subscript r stands for regular, and the diverging remainder
, where the subscript r stands for regular, and the diverging remainder
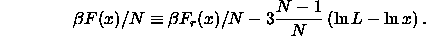
The condition for the equality of the chemical potential now becomes
where we have dropped the terms that cancel on the left and right hand
side and where we have ignored terms that vanish in the limit
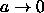 .
.
| Figure: | Scaled equations of state for hard
spherocylinder solids in the
limit 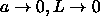 (see
eqn.
5.43). The dotted curve denotes a compression
and the solid curve corresponds to an expansion (see
eqn.
5.43). The dotted curve denotes a compression
and the solid curve corresponds to an expansion |
| Table: | Regular part of the Helmholtz free energy per particle in the close packed limit. The values have been obtained by Einstein integration. |
Figure 5.8 shows the ``equation of state'' 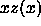 for
the dense plastic crystal and ordered solid.
The free energies of the reference systems are given in table
5.10. The coexistence values of x, pressure and
chemical potential that follow from equations 5.43 and
5.46 are
for
the dense plastic crystal and ordered solid.
The free energies of the reference systems are given in table
5.10. The coexistence values of x, pressure and
chemical potential that follow from equations 5.43 and
5.46 are
where 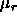 , is the regular part of the chemical potential, given by
eqn.
5.46.
As x is defined as the ratio of L and a, eqn.
5.47
describes the initial (small L) dependence of the plastic-ordered
transition on the spherocylinder length.
As presented, a given value of x corresponds to a slope of the
coexistence curve in the L,a plane.
However, by using the definition of x,
, is the regular part of the chemical potential, given by
eqn.
5.46.
As x is defined as the ratio of L and a, eqn.
5.47
describes the initial (small L) dependence of the plastic-ordered
transition on the spherocylinder length.
As presented, a given value of x corresponds to a slope of the
coexistence curve in the L,a plane.
However, by using the definition of x,

every value of x corresponds to a curve in the
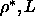 plane.
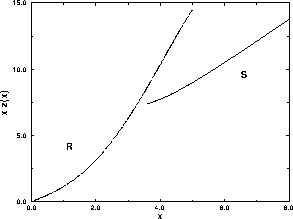
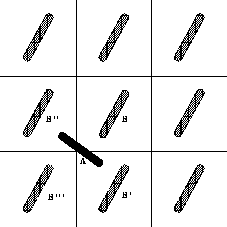
The resulting solid-solid coexistence curves are plotted
in figure 5.7. It is interesting to note that,
even at
plane.
The resulting solid-solid coexistence curves are plotted
in figure 5.7. It is interesting to note that,
even at 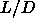 as large as 0.3,
the slope of rotator-solid coexistence curve
is still
dominated by the behavior at close packing
as large as 0.3,
the slope of rotator-solid coexistence curve
is still
dominated by the behavior at close packing