


Next: Phase diagram for Up: Simulation techniques Previous: Kappa integration
Next: Phase diagram for
Up: Simulation techniques
Previous: Kappa integration
The location of a fluid-solid
coexistence curve can be determined by performing several
free-energy calculations and measurements of the equation-of-state for
a large number of 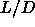 values.
However, this approach is computationally
rather expensive. To avoid this problem, we use a modification of a method that was
recently developed
by Kofke to trace coexistence curves [84]. The advantage of this method
is that only equation-of-state information at the coexistence
curve is required to follow the
values.
However, this approach is computationally
rather expensive. To avoid this problem, we use a modification of a method that was
recently developed
by Kofke to trace coexistence curves [84]. The advantage of this method
is that only equation-of-state information at the coexistence
curve is required to follow the 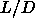 -dependence of the melting
curve. In its original form, the Kofke scheme is based on the
Clapeyron equation which describes the temperature-dependence
of the pressure at which two phases coexist (see section 4.3):
-dependence of the melting
curve. In its original form, the Kofke scheme is based on the
Clapeyron equation which describes the temperature-dependence
of the pressure at which two phases coexist (see section 4.3):
where 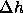 is the molar enthalpy difference and
is the molar enthalpy difference and
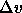 the molar volume difference of the two phases. This
equation is not self starting, in the sense that one point on the
coexistence curve must be known before the rest of the curve can be
computed by integration of eqn.
5.23. Kofke refers to
this method as ``Gibbs-Duhem'' integration.
the molar volume difference of the two phases. This
equation is not self starting, in the sense that one point on the
coexistence curve must be known before the rest of the curve can be
computed by integration of eqn.
5.23. Kofke refers to
this method as ``Gibbs-Duhem'' integration.
In the present case, we are not interested in the temperature
dependence of the coexistence curve (this dependence is trivial for
hard-core systems), but in the dependence of the coexistence pressure
on 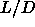 , the length-to-width ratio of the spherocylinders.
In order to obtain a Clapeyron-like equation relating the
coexistence pressure to
, the length-to-width ratio of the spherocylinders.
In order to obtain a Clapeyron-like equation relating the
coexistence pressure to 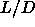 , we should first write down the explicit
dependence of the (Gibbs) free energy of the system on
, we should first write down the explicit
dependence of the (Gibbs) free energy of the system on 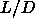 :
:

where  is the derivative
is the derivative 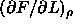 defined in eqn.
5.20 and where we have used the fact that D
is our unit of length.
Along the coexistence curve, the difference in chemical potential of
the two phases is always equal to zero. Hence,
defined in eqn.
5.20 and where we have used the fact that D
is our unit of length.
Along the coexistence curve, the difference in chemical potential of
the two phases is always equal to zero. Hence,
where 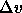 is the difference in molar volume of the two phases
at coexistence and
is the difference in molar volume of the two phases
at coexistence and 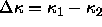 .
From eqn.
5.25 we can immediately deduce the equivalent
of the Clausius-Clapeyron equation
.
From eqn.
5.25 we can immediately deduce the equivalent
of the Clausius-Clapeyron equation
Kofke used a predictor-corrector scheme to integrate eqn. 5.23. However, we find that this integration scheme, when applied to eqn. 5.26, is not very stable. In particular, when the predicted pressure is slightly off, the predictor-corrector scheme leads to unphysical oscillations. We therefore introduce a slightly different integration procedure. Instead of calculating one new point in the phase diagram every step, we start with a set of simulations at different L values and pressure for both phases. The derivatives are calculated according to eqn. 5.26 and subsequently fitted to a polynomial in L

This polynomial is integrated to give new pressures which are used in the next iteration. The old and new pressures are mixed together to improve the stability. This procedure is repeated until convergence of the pressure has occurred. The set of starting values are obtained by application of the original Gibbs-Duhem scheme.
In Kofke's application of the Gibbs-Duhem method, the MC simulations are
carried out in the isothermal-isobaric (NPT) ensemble.
However, in the present case (hard-core particles), it is more
efficient to use Molecular Dynamics to compute the derivative
 . In practice, we use a hybrid approach where MD simulations are
embedded in a constant NPT-MC scheme. True constant-pressure MD is not an
attractive option for hard-core models.
. In practice, we use a hybrid approach where MD simulations are
embedded in a constant NPT-MC scheme. True constant-pressure MD is not an
attractive option for hard-core models.


