Trp-cage protein folding landscape
By Jarek Juraszek
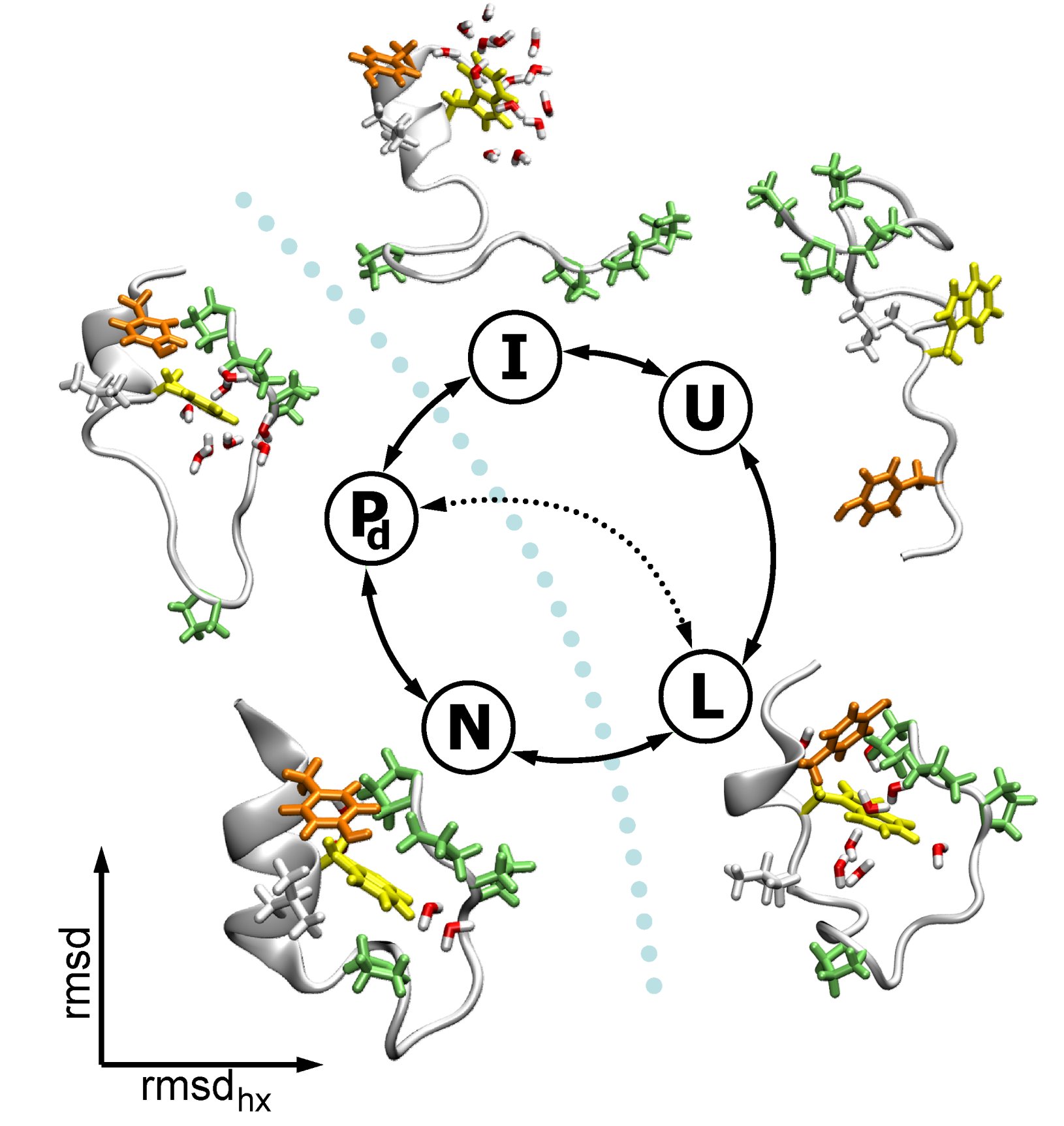
We have used Replica Exchange Molecular Dynamics (REMD) in order to investigate the free energy landscape of Trp-cage, very fast folding mini-protein. Starting from the unfolded structure we were able to sample some interesting intermediate states for the folding process, but we were not able to cross the rate limiting barrier towards the native state. For this purpose we use Transition Path Sampling (TPS), that allows us to sample unbiased dynamical pathways between the folded and intermediate states. We find that the protein follows two distinct pathways: one via forming the tertiary contacts and the salt bridge before the helix formation, the other by first forming the helix. We find the expulsion of water from the hydrophobic core as the last step upon folding.
Folding mechanism of the GB1 beta-hairpin in Trpzip4 protein
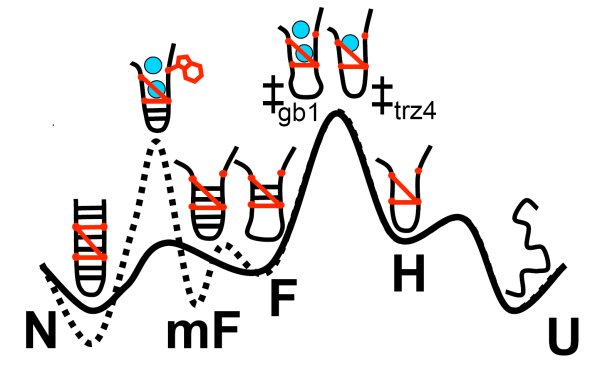
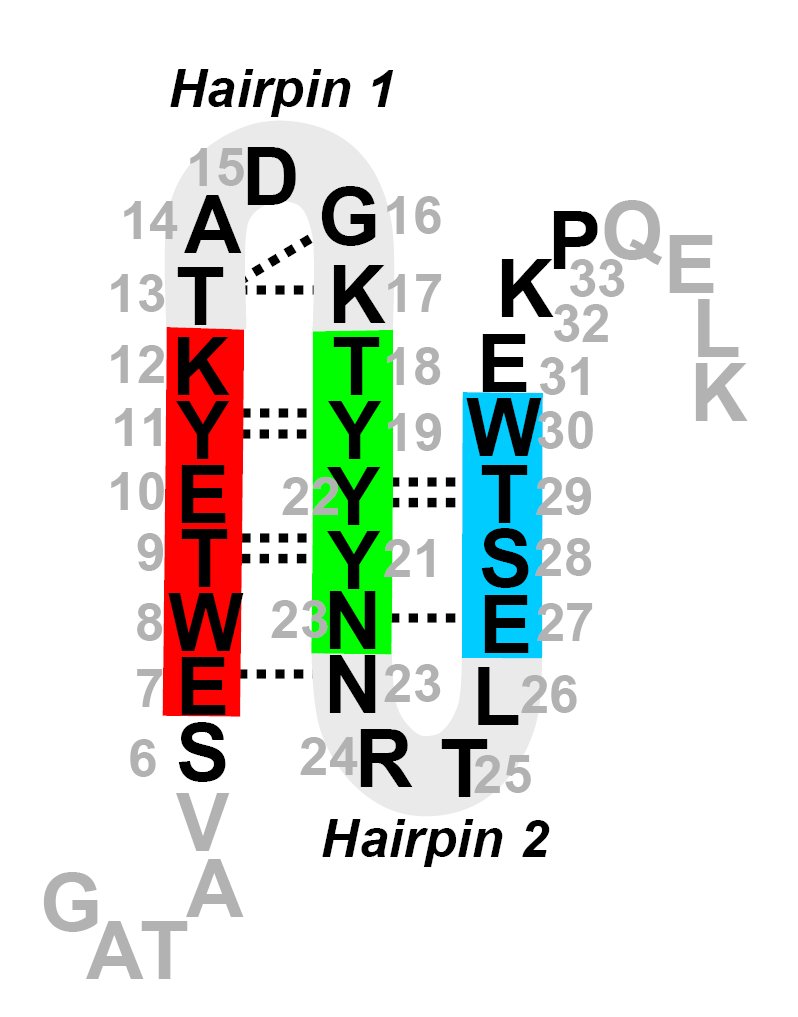
We have performed an extensive simulation study of the effects of a mutation of the GB1 beta-hairpin in Trpzip4, which has a much more stable native state, but its folding rate does not differ significantly from the original beta-hairpin. We employ Replica Exchange Molecular Dynamics to sample the free-energy landscapes of both hairpins. We attempt to identify the rate limiting barriers and sample them with the Transition Path Sampling method. We find that the increase of stability is mainly due to very strong hydrophobic interactions of the sidechains. The appearance of a misfolded state, mF, hinders the sampling in REMD. Finally we show that, mF, is an on-pathway intermediate and that the last step of folding of Trpzip4 is re-zipping and rearrangement of sidechains, even though the rate limiting barrier is the hydrophobic collapse.
Folding rate and order parameters
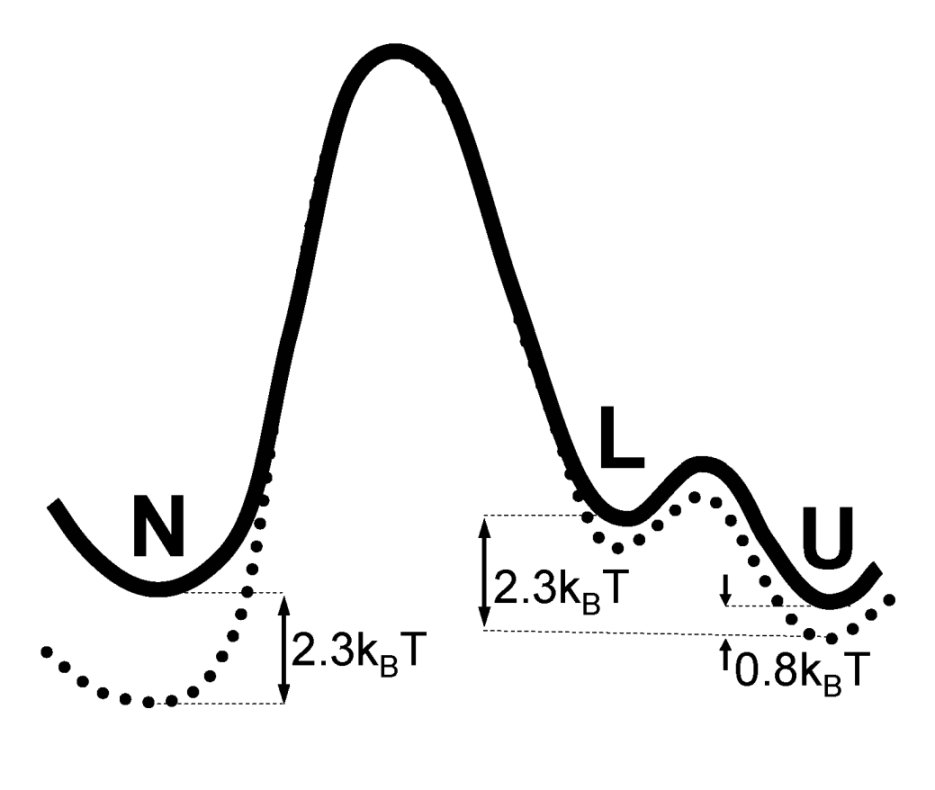
We report rate constant calculations and a reaction coordinate analysis of the rate limiting folding and unfolding process of the Trp-cage mini-protein in explicit solvent using Transition Interface Sampling (TIS). Previous Transition Path Sampling simulations revealed that in this (un)folding process, the protein maintains its compact configuration, while a (de)increase of secondary structure is observed. The calculated folding rate agrees reasonably with experiment, while the unfolding rate is 10 times higher. We discuss possible origins for this mismatch. We recomputed the rates with the Forward Flux Sampling (FFS) method, and found a discrepancy of 4 orders of magnitude, probably caused by the method's higher sensitivity to the choice of order parameter with respect to TIS. Finally, we used the previously computed TPS ensemble to screen combinations of many order parameters for the best model of the reaction coordinate by employing likelihood maximization. We found that a combination of the root mean square deviation of the helix and of the entire protein was, of the set of tried order parameters, was the one that best describes the reaction coordination.
Metadynamics simulations of WW formin binding protein 28 (FBP28)
We study the folding routes of a WW formin binding protein 28 (FBP28) at ambient conditions using advanced molecular simulation techniques. We perform Replica Exchange simulations in order to explore the unfolded state basin and possible intermediate states on the way to the folded state. Bias-Exchange Metadynamics allows unfolding the protein at room temperature and yields a number of unfolding pathways that act as initial paths for Transition Path Sampling. Subsequent equilibration of the path ensemble leads to the discovery of two major routes. Both routes pass through a close-to-native intermediate state, characterized by a detached strand B3 and Trp-30 forming non-native hydrophobic contacts. Having established good reaction coordinates, we use Metadynamics in order to calculate the corresponding barriers, and find the unfolding barrier of the order of 17 kBT, in line with the experimental values.
Conformational dynamics of the Photoactive Yellow Protein
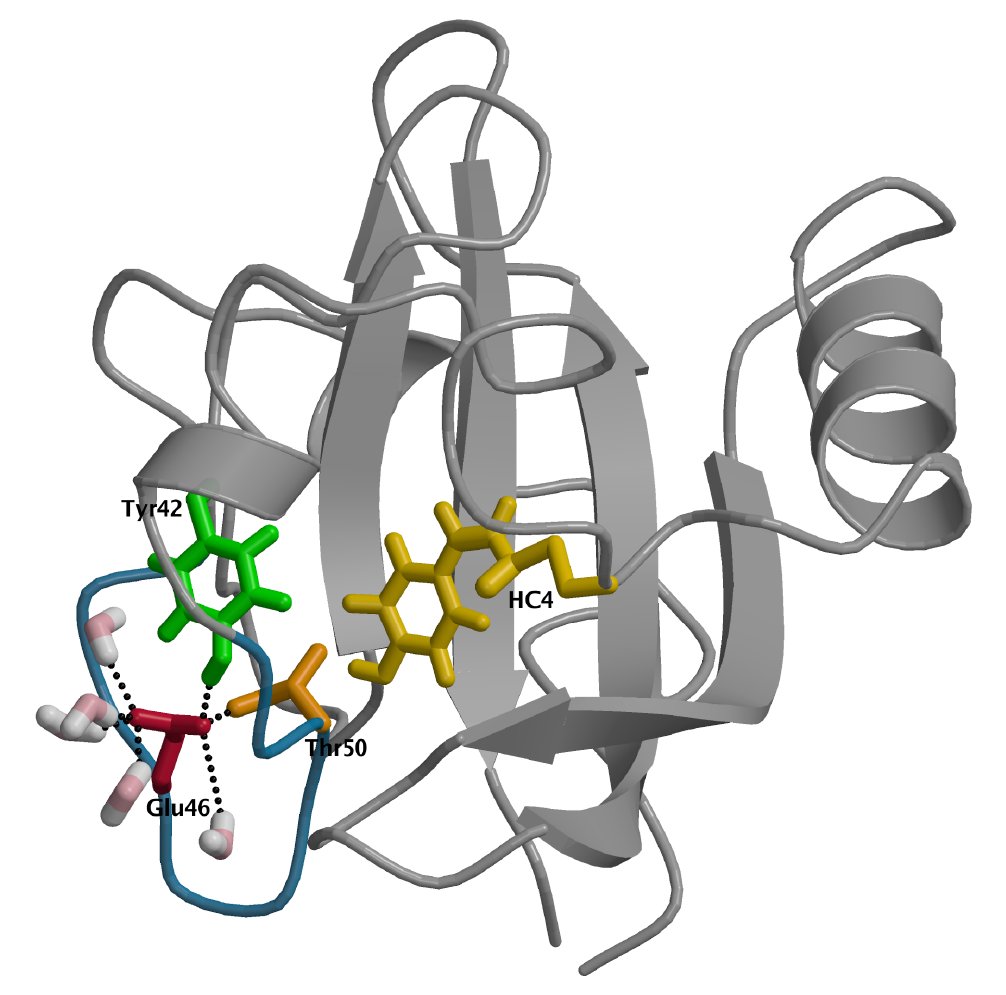
The bacterial sensor Photoactive Yellow Protein (PYP) signals the presence of blue light by undergoing a series of conformational changes. We present transition path sampling simulations of the conformational transitions that occur during the formation of the signaling state of PYP. These transitions are the unfolding of the alpha-helical region 43-52 and the solvent exposure of the chromophore and Glu46. Our simulations enable characterization of the transition states in these conformational changes. Likelihood maximization analysis further substantiates our initial description of the transition states. As a result, we present a detailed atomistic characterization of the mechanisms underlying the formation of the signaling state of PYP.