chemical reactions inside proteins
By Elske Leenders
The photocycle of the photoactive yellow protein (PYP, see signal transduction) starts with excitation, isomerisation and protonation of the chromophore, p-coumaric acid. These (chemical) reactions cannot be simulated with only conventional moleculary dynamics techniques, as they involve electron density rearrangements, bond breaking, changing partial charges on atoms and hence changing forces between particles. To simulate this properly, we need quantum chemical methods. Dynamical simulations based on quantum mechanics were first made possible by Car and Parrinello with their Car-Parrinello Molecular Dynamics. It is now the standard in the field and widely used. Recently a new algorithm was developed based on Born-Oppenheimer MD; it can handle larger systems with larger time steps and it is implemented in CP2K. Both these methods are used within the group, to simulate reactions in proteins, but also to simulate solvation of ions and small molecules (Elske Leenders and Jasper Heuft) and to simulate liquid carbon (Francesco Colonna and Luca Ghiringhelli).
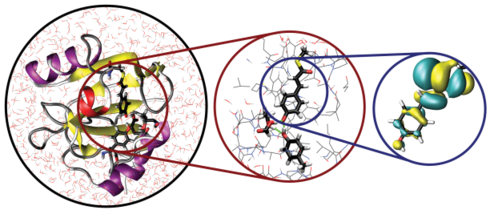
Dynamical quantum methods enable us to simulate processes ab initio and in great detail. But they do so at considerable computational costs. With the methods described above, one can simulate maybe a couple of hundreds of atoms for a couple of picoseconds. Within the length scales of proteins and the time scale of the photocycle (milliseconds), this is almost nothing. A way to overcome the size barrier is QMMM, quantum mechanics/molecular mechanics. With this method, one can use quantum chemistry on a small part of the protein were the electron density is important and (chemical) reactions happen. For the rest of the protein and the solvating water molecules, normal forcefield molecular dynamics is used. The picture provides an impression of this method. We use it to study the proton transfer to the chromophore of PYP.
Publications
- Predicting the signaling state of Photoactive Yellow Protein. J. Phys. Chem. B 111 (2007), 3765-3773 DOI: 10.1021/jp067158b