Hybrid molecular dynamics
By Bernd Ensing
Atomistic versus coarsegrained
With current highly parallelized super computers and optimized simulation software, we can perform molecular simulations in atomistic detail for systems that contain several hundreds of thousands of atoms for tens to hundreds of nanoseconds. To model even larger systems or simulate longer times, we can try to model our system at a coarsegrained resolution. Here, coarsegraining refers to lumping neighboring atoms together into single interaction sites, thus drastically reducing the number of particles and interactions in the calculations. Coarsegraining is thus much less computationally demanding, but at the cost of giving up on the atomistic details (click here for an example of a coarsegrained simulation).
Multiscale simulation
 A box of methane molecules treated atomistically within a
spherical region and coarsegrained outside
A box of methane molecules treated atomistically within a
spherical region and coarsegrained outside
Hybrid Molecular Dynamics (hybrid MD) is a multiscale technique that combines the advantages of both atomistic and coarsegrained MD: low computational cost which allows for extended systems and timescales plus atomistic detail in specific regions where necessary.
Examples of applications where a multiscalle apporach would be beneficial could be: proteins (atomistically) in solvent (coarsegrained), adsorption or growth on surfaces (treating only the interface region in atomistic detail), or diffusion of a particle through a complex fluid (using a spherical atomic region centered on the particle).
Adaptive resolution
The coupling between particles in the different resolution regions can simply be done at the coarsegrained interaction level. This entailes just computing the positions in the coarsegrained representation of the particles in the atomistic region, then evaluation the forces using the coarsegrained forcefield and finally distributing the coarsegained force over the atoms in the atomstic region.
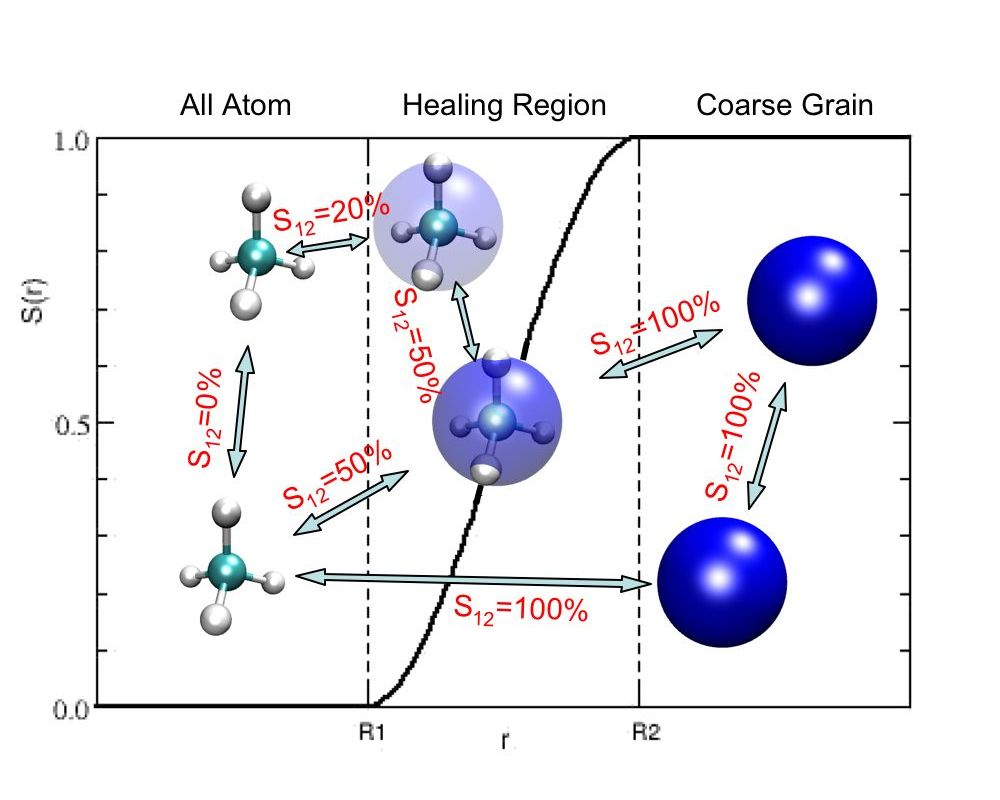
Each methane molecule is attributed a value for its amount of coarsegrained character (s(r)). The interaction potential between particles is weighted by the maximum s(r) among the interacting particles.
A problem is however the treatment of particles that cross a boundary between regions of different resolution: how does a coarsegrained particle suddenly transform into its atomistic counterpart?
This is done, by introducing an intermediate or healing region in which the particles change their representation in a smooth manner. The interaction between particles is taken to be the superposition of the atomistic a coarsegrained potentials, where the weight is determined by the position of the interacting particles as illustrated by the figure.
Constant energy
As particles adapt their representation on the fly as they move from low to high-resolution regions, and vice versa, the number of degrees of freedom changes continuously. However, using our potential scaling approach, we can nevertheless recover the total energy as a conserved quantity. This is particularly important in order to assess the quality of the integration of the equations of motion. In particular, it allows us to determine the minimal width of the healing region for a smooth resolution adaptation and to set the MD timesteps. We use the reversible RESPA multi-timestep algorithm, allowing for different timesteps in the atomistic and the coarsegrained regions, rendering the hybrid MD effectively not only a spatial, but also a temporal multiscale method.
See also: the illustrative movie on our gallery webpage.
Publications
- Recent progress in multiscale molecular dynamics simulation of soft matter. Phys. Chem. Chem. Phys. 12 (2010), 12401 - 12412 DOI: 10.1039/c004111d
- Adaptive multiscale molecular dynamics of macromolecular fluids. Phys. Rev. Lett. 105 (2010), 237802 DOI: 10.1103/PhysRevLett.105.237802
- Smooth capping of short-range repulsive forces in hybrid atomistic/coarse-grain molecular dynamics simulation. Proceedings of Multiscale Materials Modeling in press (2010)
- Multiscale molecular dynamics and the reverse mapping problem. Book chapter in Trends in Computational Nanomechanics (Challenges and Advances in Computational Chemistry and Physics, Volume 9) edited by T. Dumitrica (Springer, 2010), p. 25 - 60 DOI: 10.1007/978-1-4020-9785-0_2
- Energy conservation in adaptive hybrid atomistic/coarse-grain molecular dynamics. J. Chem. Theor. Comp. 3 (2007), 1100 - 1105, DOI: 10.1021/ct600323n
- Coarse grained-to-atomistic mapping algorithm: a tool for multiscale simulations. Book chapter in Multiscale Simulation Methods for Nanomaterials, R. B. Ross and S. Mohanty, Editors. 2007, John Wiley & Sons, Inc., Hoboken, NJ, USA DOI: 10.1002/9780470191675.ch5